PDFguiの使い方(簡易マニュアル)¶
樹神さんのマニュアルを移植。
インストール (recommended)¶
PDFgui は、Anaconda Python のソフトウェア パッケージとして配布されています(2024.2.26)。
PDFgui インストールの前提条件として、Miniconda または Anaconda Python が必要です。PDFgui は、Anaconda パッケージの「conda-forge」チャネルから入手できます。これを行うには、ターミナル ウィンドウまたは Anaconda コマンドプロンプトを開き、次のように conda コマンドを使用します。
conda create --name=pdfgui2 python=3.9
conda activate pdfgui2
conda install -c conda-forge diffpy.pdfgui
PDFgui は、ターミナルウィンドウまたは Anaconda コマンドプロンプトから次のように入力して起動できます。
pdfgui
インストール(インストーラから)¶
LANSCEグループの プログラムダウンロードページ から、diffpy-1.0-r3067.exe (←最新版)をダウンロード。これを実行することによって、フォルダ
C:\DiffPy
内に必要なファイルがインストールされる。またデスクトップ上にPDFguiのショートカットが作成される。
Linux、Mac、Unixの場合には、diffpy-1.0-r3067.tgzをダウンロード。
Python上で
easy_install diffpy.pdfgui
とコマンド入力してインストール。詳細は以下のページを参照してください。
構造モデルおよび測定データの入力¶
tutorial としてNiのPDFを取り上げる。
ショートカットPDFguiをダブルクリックしてプログラムを立ち上げる。
Menu Barの「Fits」をクリックして、「New Fit」を選択。
構造モデルの入力¶
Menu Barの「Phases」をクリックして、「New Phase」を選択。
すでに構造モデルファイル“.stru”が存在する場合には、「Load a structure from file」の「Open」をクリックし、
C:\DiffPy\Python25\Lib\site-packages\diffpy.pdfgui-1.0_r3067_20090410-py2.5.egg\doc\tutorial
フォルダ中からNi.struを選択(図1の状態になる)。
新しく入力する場合には、 「Create a structure from scratch」の「New」をクリック。 「Phase Configure」パネルが図1の状態になるように各パラメータを入力。
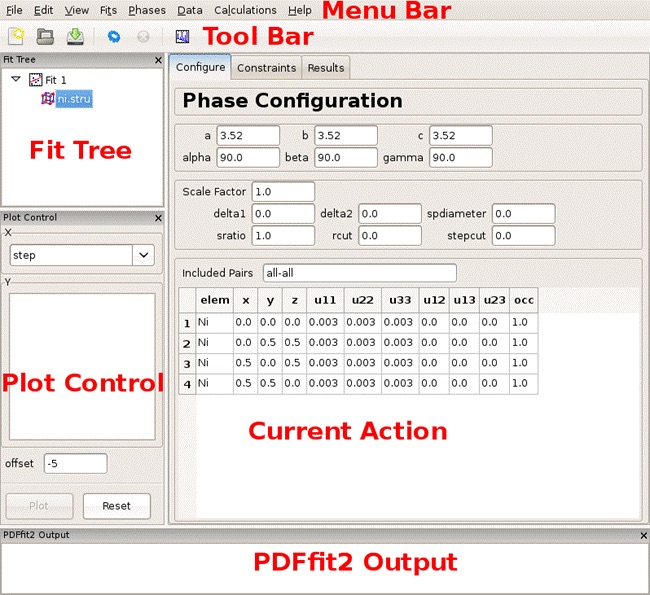
図 1 :Niの構造モデルの入力が完了¶
PDFguiでは、ユニットセル中にあるすべての原子を入力する必要がある。空間群を利用することによって、結晶学的に等価なサイトを一度に入力できる。
「elem」を右クリックして、「Inset atoms」を選択。「OK」をクリック。
1のelemをNiに変更。
Niを右クリック。「Expand space group…」を選択。
「Space Group」で 「Fm-3m」を選択。「Origin Offset」は「0.0 0.0 0.0」のまま。
OKをクリックすると、4つの等価なNiサイトが入力される(図1の状態に)。
構造モデル中の各パラメータの意味(2段目のブロック)
Scale Factor |
複数の相が存在する場合、その相についてスケールを合わせる。1相で解析する場合には測定データのScale Factorと同じなので1.0で固定する。 |
delta1, delta2 |
ピーク幅を決定する。詳細はQbroadのところで。 |
spdiameter |
粒子の直径。粒子サイズによる強度の減衰を取り入れた解析に用いる。バルク物質で強度の減衰を無視する場合にはゼロを入力。 |
sratio, rcut |
デフォルトではそれぞれ1.0, 0.0を入力。rcut以下のrにあるピークの幅をsratioだけシャープにする(Qbroadを参照)。 |
stepcut |
このrの値より先のG(r)をゼロにする。0.0を入力すると機能しない。 |
測定データの入力¶
Menu Barの「Data」をクリックして、「New Data Set」を選択。
「Load a structure from file」の「Open」をクリックし、
C:\DiffPy\Python25\Lib\site-packages\diffpy.pdfgui-1.0_r3067_20090410-py2.5.egg\doc\tutorial
フォルダ中からNi-neutron.grを選択(図2の状態になる。ただし図2ではNi-xray.grを使用しているので、いくつか異なる点があることに注意)。
各パラメータの意味
Data Range |
入力されたデータのr範囲。自動入力。 |
Fit Range |
フィットを行うr範囲 |
Scale Factor |
フィットの際にスケールを調整(理想的には1.0だが、、、)。 |
Qmax |
G(r)の導出に行うフーリエ変換のQの最大値。振動成分の周期を決める。PDFgetNで得られたG(r)では自動入力。 |
Qdamp |
Q分解能によるPDF強度の減衰を表わすGaussianの係数。exp[-(r Qdamp)2/2] |
Qbroad |
ピーク幅を決めるパラメータ。ピーク幅は以下の式で与えられる。
  for r<rcut, =1.0 for r>=rcut for r<rcut, =1.0 for r>=rcutσ’:温度因子から計算される幅。
δ1, δ1:delta1, delta2。
|
Temperature, Doping |
入力ファイルに記載されている場合に読み込まれる。解析の際には特に使用しない。 |

図 2 :NiのPDF(測定データ)の読み込みが完了¶
データ解析¶
フィッティングパラメータの設定¶
構造モデルおよび測定データ(PDF)の「Constraints」パネルでrefineするパラメータを設定する。
測定データのパラメータの設定
refineするパラメータを@1, @2...で指定。ここではScale FactorとQdampをrefineする(図3)。
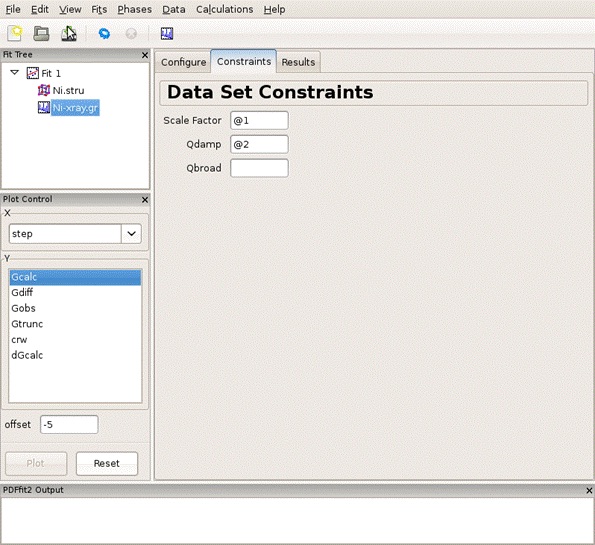
図 3 :測定データのrefineするパラメータを指定¶
構造モデルのパラメータの設定
格子定数と温度因子をrefine。温度因子は等方的とする(図4)。
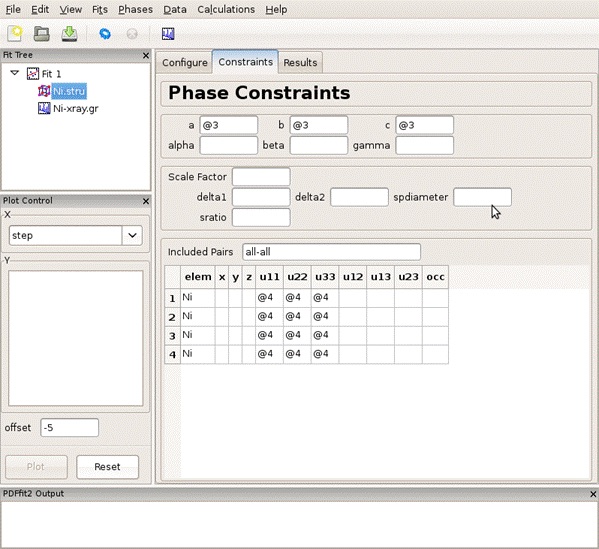
図 4 :構造モデルのrefineするパラメータを指定¶
フィッティングの開始¶
「Fit 1」をクリックすると、refineするパラメータとその初期値が表示される。
このパネルで初期値の入力やパラメータのrefine⇔fixの設定が可能(図5)。
「Tool Bar」の「Start a fit or calculation」(図5を参照)をクリックすると、フィッティングを開始する。
エラーメッセージが出たり、R-因子が小さくならない場合は、初期値を変えたり、refineするパラメータの数を減らしてみる。
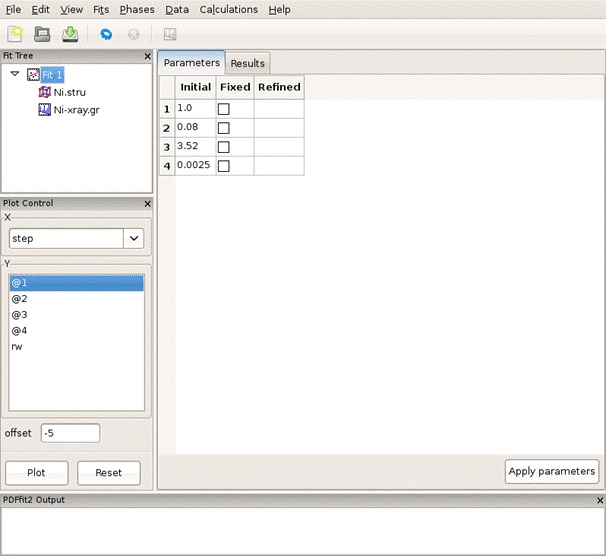
図 5 :フィッティングパラメータの初期値およびrefineの有無を設定。¶
解析結果の確認¶
フィッティングが終了すると、refineされたパラメータの値が「Refined」に表示される。
「PDFfit2 Output」にR-因子の値が表示(図6)。
「Results」をクリックすると詳しい解析結果が表示される。

図 6 :フィッティング終了後の画面。¶
フィッティング結果のグラフ表示
「Ni-neutron.gr」をクリック。
「Plot Control」でx軸にr、 y軸にGcal、Gobdを選択。
Ctrlキーを押しながらクリックするとプロットするデータを複数選択できる。
「Plot」をクリックすると結果をグラフで表示(図7)。
結果をテキストファイルに保存する場合
図7赤丸で示した「Export plot data」をクリックすると、rとGobs、rとGcalの列がテキストファイルとして保存される。
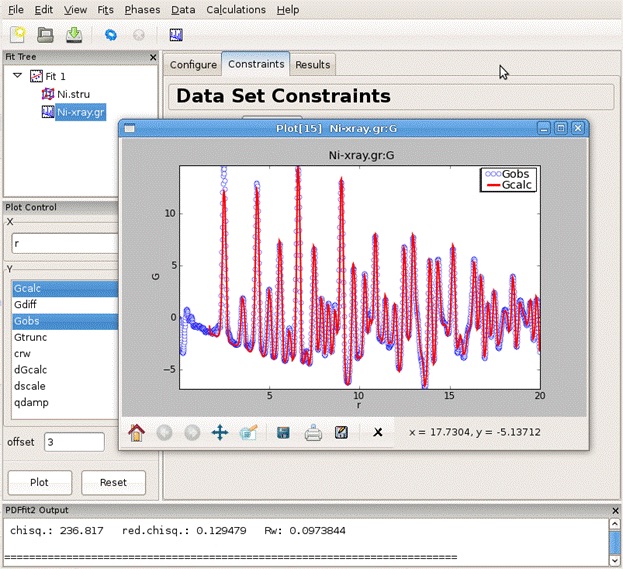
図 7 :解析結果をグラフで表示。¶
NOVAで得られたアルミナ(Al2O3)データのPDF解析¶
Niの原子位置はすべて特殊位置なので、原子位置のrefinementをしていない。そこで原子位置をrefineする例としてアルミナのデータ解析を取り上げる。
構造モデルの入力¶
Al2O3は空間群R-3c (No. 167) HEXAGONAL AXES、格子定数:a=4.75909, c=12.9918Å, γ=120°、原子位置は、Al:12c (0, 0, 0.3523) O:18e (0.30632, 0, 1/4)
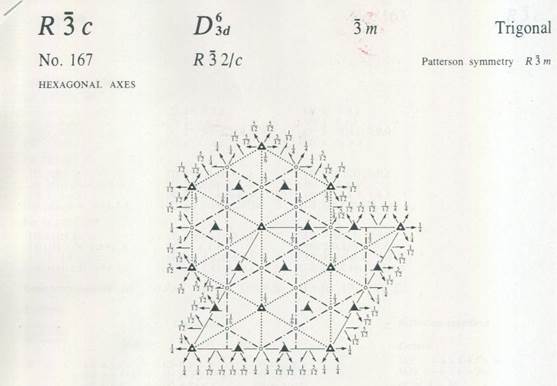
図 8 :フィッティング終了後の画面。¶
原子位置の入力
まず「Insert atoms」でAlをひとつ入力。座標は(0, 0, 0.3523)。
「Expand space group」でH-3cを選択。Alの結晶学的に等価な12サイトの座標が入力される
12番目のAlを右クリックで「Insert atoms」。O:(0.30632, 0, 1/4)を入力。
Alと同様に「Expand space group」でH-3cを選択。Oの結晶学的に等価な18サイトの座標が入力される。

図 9 :フィッティング終了後の画面。¶
入力された原子位置は、例えばAlの場合、(0, 0, z), (0, 0, -z+1/2), (0, 0, -z), (0, 0, z+1/2)、次に左の4つの座標に+(2/3, 1/3, 1/3)足したもの、次に+(1/3, 2/3, 2/3)を足したものの順に表示される(図9のInternational Tableを参照)。
測定データの入力¶
Al2O3.grを読み込む
r<1.0に大きな振動成分があるので「Fit Range」は1.0~20Åに設定。
フィッティングパラメータの設定¶
Niのときと同様に、Scale Factor等を@1, @2….でフィッティングパラメータに設定する。
ここでは原子位置のフィッティングパラメータの設定について説明。
図9のInternational Tableに示すように、Alの場合にはz位置、Oの場合にはx, y位置がフィッティングパラメータとなる。これを入力したすべての原子に対して設定する必要がある。
例えば1番目のAlについて、z位置を@8として設定した場合、2番目のAlは1つめの位置(0, 0, z)に対して(0, 0, -z+1/2)の位置にあるので、z位置の設定は -@8+1/2となる。
5番目のAlは1つめの位置 (0, 0, z)に対して+(2/3, 1/3, 1/3)の位置にあるので、z位置の設定は @8+1/3となる。
1番目のOについて、x位置を@9として設定した場合、2番目のAlは1つめの位置(x, 0, 1/4)に対して(0, x, 1/4)の位置にあるので、 y位置の設定は @9+1/4となる。
7番目のAlは1つめの位置 (x, 0, 1/4)に対して+(2/3, 1/3, 1/3)の位置にあるので、x位置の設定は @9+2/3となる。
すべてのAlおよびOについて、次ページのように設定することになる。
Elem x y z
Al @8
Al 0.5-@8
Al 1-@8
Al 0.5+@8
Al @8+0.333333
Al 0.5-@8+0.333333
Al 1-@8+0.3333333
Al 0.5+@8+0.333333
Al @8+0.666667
Al 0.5-@8+0.666667
Al -@8+0.666667
Al 0.5+@8+0.666667
O @9
O @9
O -@9 -@9
O -@9
O -@9
O @9 @9
O @9+0.666667
O @9+0.333333
O -@9+0.666667 -@9+0.333333
O -@9+0.666667
O -@9+0.333333
O @9+0.666667 @9+0.333333
O @9+0.333333
O @9+0.666667
O -@9+0.333333 -@9+0.666667
O -@9+0.333333
O -@9+0.666667
O @9+0.333333 @9+0.666667