原子間ポテンシャル¶
原子間ポテンシャルは、2つの原子を近づけていくと、ある距離までエネルギーが減少する。これは2つの原子に電子が共有されることが原因である。一方で近づきすぎると、電子同士の反発する力が強くなるためエネルギーは急上昇する。
このような原子間ポテンシャルは、第一原理計算と呼ばれる計算手法によって求めることが可能である。ここでは、Materials Projectに収録されている計算データを利用する。
Jupyter notebookを使用して、FCC構造のCuについて原子間ポテンシャル(全エネルギーと体積との関係)を図示してみましょう。
[1]:
from pylab import * #this includes numpy as np!
from scipy.optimize import leastsq
plt.rcParams["figure.figsize"] = (5,4)
FCC構造のCuの計算データはmp-30として登録されている。体積をv、エネルギーをeとします。またvを原子間距離のrにするため、np.cbrt(v)としてvの立方根をとる。
[2]:
#Cu(mp-30)
v = np.array([8.6664, 8.9586, 9.5623, 9.8741, 10.5179, 9.2571, 10.8500, 11.5351, 12.9905, 12.2484, 13.7620, 12.6158, 14.5635, 11.1890, 14.9757, 15.8231, 10.1926, 15.3955, 11.8882, 14.1590, 13.3726])
r = np.cbrt(v)
e = np.array([-3.3243, -3.4941, -3.7562, -3.8536, -3.9921, -3.6372, -4.0371, -4.0873, -4.0660, -4.0937, -4.0115, -4.0836, -3.9364, -4.0681, -3.8927, -3.7961, -3.9314, -3.8457, -4.0956, -3.9763, -4.0416])
plot(r,e,'ro')
xlabel('r ($\AA$)')
ylabel('Energy (eV)')
[2]:
Text(0, 0.5, 'Energy (eV)')
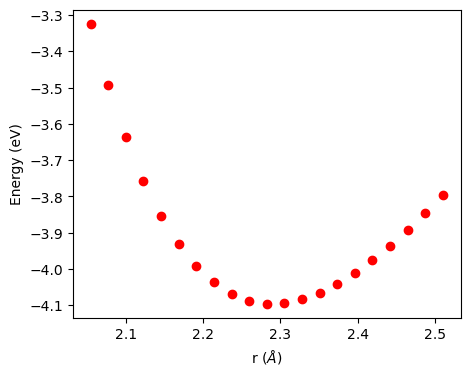
原子間ポテンシャルは\(r_0 \sim 2.3\) の周りで極小値を取ることがわかる。
二次関数でフィッティング¶
この原子間ポテンシャルはどのような関数で表現されるのであろうか。原子間に働く相互作用を2つの球体を繋ぐバネのようなもの(調和振動子)で考えた場合、原子間ポテンシャルは原子間距離\(r\)の2乗に比例する。
そこで二次関数 \(ar^2 + br + c\)でフィッティングしてみる。
[3]:
#make a vector to evaluate fits on with a lot of points so it looks smooth
rfit = np.linspace(min(r),max(r),100)
a,b,c = polyfit(r,e,2) #this is from pylab
plot(r,e,'ro')
plot(rfit, a*rfit**2 + b*rfit + c,'--',label='parabolic fit')
xlabel('r ($\AA$)')
ylabel('Energy (eV)')
[3]:
Text(0, 0.5, 'Energy (eV)')
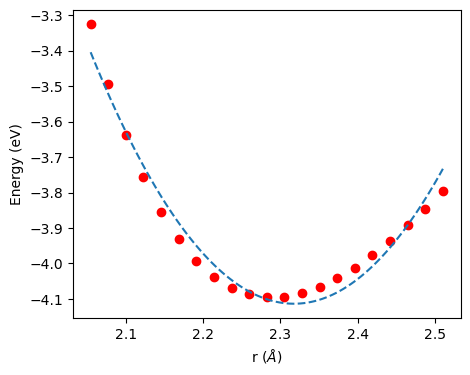
2次関数を用いたフィッティング結果をみると、平衡原子間距離 \(r_0 \sim 2.3\) を中心にして、赤丸のデータは対象な形をとっていないことがわかる。
赤丸の計算値は、右側にやや平らな形状をしている。これは原子間ポテンシャルが調和振動子では記述しきれない非調和性を持っていることを示している。熱膨張はこのような原子間ポテンシャルの非対称性(非調和性)によって現れる。
温度が上昇すると平衡原子間距離 \(r_0 \sim 2.3\) から振動するのであるが、
rが小さい領域のポテンシャルは急峻になっている
rが大きい領域のポテンシャルはなだらかになっている
これによってrが大きい領域へ平均原子間距離が動き、熱膨張を生じるということになる。
Morse ポテンシャルでフィッティング¶
この様な固体のミクロの振舞いを記述するモデルを考える。固体中の原子間に働く力を数学的に記述する2体間ポテンシャル (pair potential) にはさまざまなものがあるが,簡単なものとしてモース (Morse) 型がある。
Morseポテンシャルは,2原子分子の原子間距離\(r\)に依存するポテンシャルである.
ここで\(r\)は原子間距離。\(r_0\)は平衡原子間距離。\(A\)、\(D\)、\(\lambda\)はフィッティングパラメータである。Cuのエネルギーと体積の関係に、モース型ポテンシャルをフィッティングしてみよう。
*****here*****
に正しい式を記入して実行してください。
[4]:
#difinition of the functions
def Morse(parameters,r):
A = parameters[0]
D = parameters[1]
r0 = parameters[2]
la = parameters[3]
*****here*****
return E
def objective(pars,y,x):
#we will minimize this function
err = y - Morse(pars,x)
return err
[ ]:
#now here are our initial guesses.
A = -4.1
D = 1
r0 = 2.3
la = 0.1
x0 = [A, D, r0, la]
morpars, ier = leastsq(objective, x0, args=(e,r)) #this is from scipy
[ ]:
plot(r,e,'ro')
plot(rfit, Morse(morpars,rfit), label='Morse fit')
xlabel('r ($\AA$)')
ylabel('Energy (eV)')
legend(loc='best')
#add some text to the figure in figure coordinates
ax = gca()
text(0.4,0.5,'A = %1.2f eV' % morpars[0],transform = ax.transAxes)
text(0.4,0.4,'D = %1.2f eV' % morpars[1],transform = ax.transAxes)
text(0.4,0.3,'r0 = %1.2f $\AA$' % morpars[2],transform = ax.transAxes)
text(0.4,0.2,'lambda = %1.2f $\AA^{-1}$' % morpars[3],transform = ax.transAxes)
savefig('a-eos.png')
show()
正しい結果は以下のようになります。
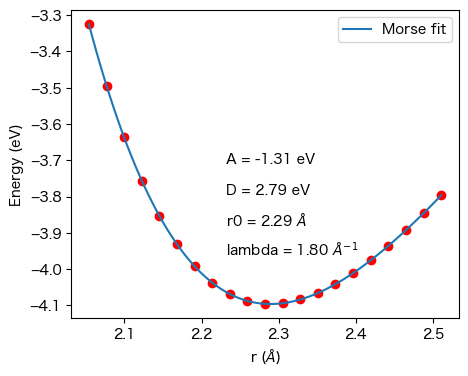
二次関数の時よりもフィッティング結果が良好であることがわかる。ここで、
\(r_0\)の平衡原子間距離は2.29 Å
\(A\)=-1.31 eV、\(D\)=2.79 eV、\(\lambda\)=1.80 Å\(^{-1}\)
である。
課題¶
The Materials Projectからダウンロードした原子間ポテンシャルのデータをMoodleからダウンロードしてください(EOS.xlsx)。Jupyter notebookを使用して、これらのデータを読み込み、各元素の原子間ポテンシャルのグラフを作成しましょう。その結果から、それぞれの原子間の安定な原子間距離と最安定なエネルギーを求めてください。
提出ファイルはJupyter notebook。