結晶構造¶
原子間力¶
原子は互いに引きあって液体や固体をつくる.原子の種類や組合せによって,その引きあう力,化学結合の性質が異なる.ヘリウムやアルゴンのような希ガスではその結合力は小さく,なかなか液体や固体にならない.一方,多くの金属や化合物は強い結合力をもち,高温まで安定である.原子間の強い結合は, イオン結合 , 共有結合 , 金属結合 に分類される.
イオン結合¶
イオン結合(ionic bond)は,たとえば食塩(NaCl)中のNa+とCl-のように,正負のイオンが 静電引力(クーロン引力) を及ぼしあい,結合するものである.その結晶構造は NaCl型 のようである.同じイオン結合でも塩化セシウム(CsCl)の結晶構造は CsCl型 のようになる.
イオン結晶の構造:NaCl型ナトリウム原子は,原子番号が11であり,K殻(1s2)とL殻(2s22p6)の外側の不安定な3s電子を放出して1価陽イオンNa+になることにより安定する.セシウム原子(原子番号 55) も同様にCs+になりやすい.一方,塩素原子(原子番号 17) は,1つ電子をもらって 1 価陰イオンCl-になることにより電子配置が1s22s22p63s23p6となり安定する.電子配置についてその本質を理解するには量子力学の知識が必要である(金属材料学II).
K殻,L殻などの閉殻構造より電子が1つ多いIA 族元素 Li, Na, K, Csなど,2つ多い II A族元素Be, Mg, Caなどは陽イオンになりやすい.逆に,閉殻構造より電子が1つ足りないVII A族元素 F, Cl, Brや2つ足りないVI A族元素 O, S, Seなどは陰イオンになりやすい.イオン結合の強さ(安定性)は,イオンになることの容易さ,すなわち正イオンならば 電気的陽性 (electro-positive)の度合い,負イオンならば 電気的陰性 (electro- negative)の度合いによってほぼ決まる.たとえば食塩の結合エネルギーは約 800 kJ/molである.
共有結合¶
共有結合(covalent bond)は2つの原子が電子を共有することによって安定な電子配置をつくったものである.たとえば水素はH2, フッ素はF2, 塩素Cl2なる二原子分子をつくるが,この際,相互に1つずつの電子を共有しあい,見かけ上8個(水素は2個)の電子をもつ擬似閉殻構造となる.その様子をH : H, F : F, Cl : Clのように表している.:の記号は2つの共有された電子を表す.
IV A族元素である炭素は,4つの電子を隣の原子と共有しあって共有結合結晶ダイヤモンドをつくる.その結晶構造を ダイヤモンドの結晶構造 に示す.ある炭素原子からみて隣の4つの原子が正四面体を構成するように,結合の手が伸びている.このように共有結合では,結合が特定の方向を向いており,次に述べる金属結合との大きな違いである.周期律表上同族のケイ素(シリコン),ゲルマニウムも同じくダイヤモンド構造の結晶をつくる.
V族,VI 族の元素も1原子当たりそれぞれ3つないし2つの共有結合をつくって空間的に拡がった共有結合結晶をつくる.族の数をNとすると結合手の数は8 – Nである.VII 族元素は結合手が1つしかないため,前述のように閉じた二原子分子になる.そのほか,炭索と水素の化合物メタン(CH4),エタン (C2H6),エチレン (C2H4),アセチレン(C2H2)などの炭化水素も共有結合によってつくられた分子である.
ダイヤモンドの結晶構造金属結合¶
ナトリウム金属は金属結合によって結びついた典型的な金属結晶である.原子1つ当たり1つずつ放出された電子は,1つのイオンのまわりに束縛されることなく,結晶全体を動きまわる.このような電子を 遍歴電子 (itinerant electron)と呼ぶ.また,この電子が結晶中を移動することによって,電気や熱を運ぶので 伝導電子 (conduction electron)ともいう.
金属結合は 図 1 に示すように,遍歴する電子が正イオン同士を結びつけており,結合に特定の方向はなく角度依存性がない.このため,金属結晶では,パチンコ球のような丸い原子というイメージが適切である.
周期律表をみるとわかるように,元素が単体で (1種類の原子で)凝集すると,多くの場合(92元素中60以上)金属になる.周期律表上のIV A ないしVIII A族の間の元素を除いて,おおむね金属である.また,金属元素と金属元素よりなる物質,すなわち合金もまた, 金属的な結合状態を示す.しかし,金属とI V AないしVIII A族元素よりなる化合物では,イオン結合性や共有結合性が強くなる.
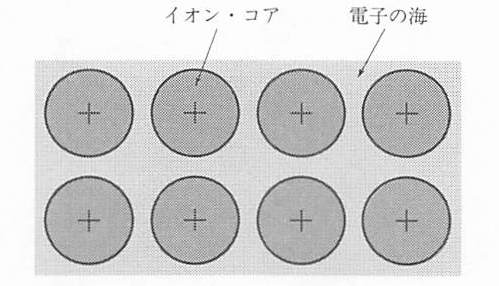
図 1 金属結合の概念図¶
以上述べたイオン結合,共有結合,金属結合は概念的なものであって,さまざまな物質が明確に区分されるものでないことを注意しておこう.化合物がイオン結合性と共有結合性をあわせもつことは粋通である(物質ごとにその比率が求められている).また,金属結合といえども共有結合的な性質を含んでいるし,2種類の元素よりなる合金においては,その電気陰性度の違いによって,イオン結合的な電荷分布の偏りが生じる.
ファン・デル・ワールス結合¶
ヘリウムやネオン,アルゴンは電子配置が閉殻構造をなしているので,上に述べた原子間力は働かない.しかし,原子内の電子位置が時間変化するので,原子核に対し,瞬間的に電子分布の偏りができ,電気的分極を生じる.この電気的双極子が引きあうことにより弱い結合が生じる.これが ファン・デル・ワールス力 と呼ばれるものである.この結合エネルギーは数 kJ/mol程度で,NaClの100分の1程度にすぎない.
二体原子間ポテンシャル¶
Na+イオンに無限大の距離からCl-イオンを近づけてくる場合を考えよう.正負イオンが 距離rにあるときのクーロン引力の大きさは \(e^2/r^2\) である.eはイオンの電荷である.無限遠から距離rまで近づけたときの位置エネルギーの変化は
Vは負であり,近づけることによりエネルギーが下がることを示している.一方距離rが小さくなると,閉殻を構成する電子同士が接触するようになり,反発力が生じる.この様子を 図 2 に示す.この反発力は距離とともに急速に増大する.両者が釣り合うところが安定位置であり,原子間距離r0を与える.正イオンの半径をr+, 負イオンの半径をr-とすれば
である. またr=r0とr= \(\infty\) との位置エネルギーの差が結合エネルギーV0である.
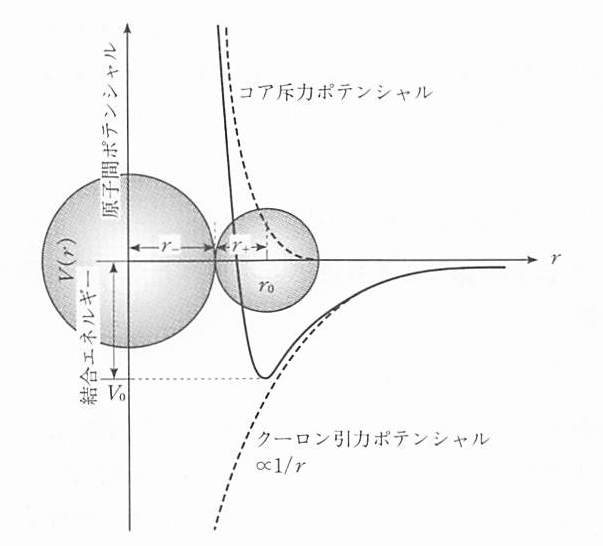
図 2 原子間ポテンシャルの距離による変化(イオン結晶の場合).最も低い位置が原子間距離になる.¶
以上は,イオン結合する2つの原子間の力を考えたものであるが,金属結合や共有結合の場合にも,ポテンシャルの形(図中の曲線の形)こそ違うが,似たような曲線が得られる.固体中では多数の原子との相互作用を考える必要があるが,それらを組み入れて,2つの原子間の相互作用として近似的に表すことができる.そのようにして原子間相互作用を2つの原子間の距離の関数として表したものを 二体原子間相互作用エネルギー (pairwise interaction)または 二体原子間ボテンシャル という.
二体原子間ポテンシャルが与えられれば,固体がどれくらい硬いか, 熱膨張の大きさはどのくらいかが求められる.原子間距離を平衡位置r0からわずかに変位させてrにもってくるとすると,そのときの復元力は \(F=-\partial V /\partial r\) である.
いま,外力F'が与えられたとすると,F' + F = 0となるように原子が変位して釣り合おうとする.ポテンシャルVをr = r0のまわりでテイラー展開すると
ここでr = r0では \(\partial V/\partial r=0\) であることを用いた.高次の項を省略し,式 (2) をrで微分して,
変位r – r0は外力F'に比例し,その比例係数は \((\partial^2 V/\partial r^2)_{r=r_0}\) である.外力によって固体中のすべての原子について原子間隔が一様に伸縮すると考えれば比例定数 \((\partial^2 V/\partial r^2)_{r=r_0}\) すなわち曲線のくぼみの曲率が 弾性定数 を与える.
次に熱膨張について考えよう.温度が上昇すると原子は熱エネルギーによって,その平衡位置を中心に熱振動する.このとき原子の獲得する平均のエネルギーは,1つの振動方向につきkBTであり, 図 3 に示すように,ポテンシャルの底から高さkBTの範囲で原子位置は変化する.ここでkBはボルツマン定数,Tは絶対温度である.
ポテンシャルが式 (2) に示すような2次曲線であれば,変位が対称的なので熱膨張は起こらないが,実際は高次の項があり,rの大きい側でなだらかになっているので,温度とともに平均の原子位置が外側へずれる.ポテンシャルの形が広く浅いと温度による非対称変位が大きく現れ,熱膨張率(係数)が大きくなる.熱膨張率αは,温度1℃の上昇に対する長さの変化率Δl/lで表され,金属では10-5程度,ダイヤモンドでは1桁小さく1.2×10-6である.プラスチックは10-4程度と大きい.
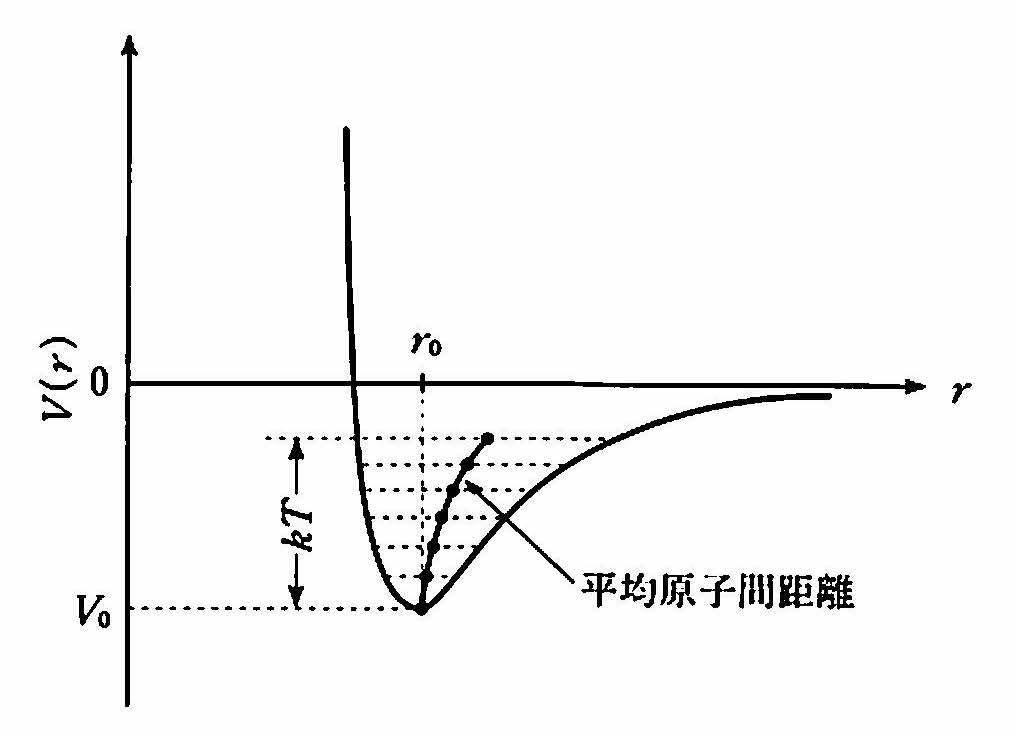
図 3 熱振動(格子振動)による平均原子間距雄の増大が熱膨張を引き起こす.¶
結晶構造¶
結晶の定義¶
すべての固体は 結晶 か, 非結晶(アモルファス固体) か 準結晶 に分類される.準結晶は,1984年にその存在が明らかになった新しい物質である.
原子が規則正しく周期的に配列したものを結晶(crystal)という.クリスタルとはもともと水晶を指す言葉で,定まった面で形づくられた水晶の美しい外形から,内部の規則正しい原子配列が早くから予想されていた.20世紀初頭になってその実際の原子配列が X線回折法 によって求められるようになった.
さて,配列の周期性とは,ある任意の位置rから距離R離れた位置r + Rでの状況がもとの位置rでの状況と全く等しいことをいう.ここでRは,3次元空間では周期を表す3つの独立なベクトルa, b, c を用いて,
と表せる.全く同じつくりの団地では,各家の風呂場や台所,居間は同じ位置にあり,その位置を家の隅から測ってrとすれば,周期aずらした隣の家でも,また,たて,横方向にRずらした家でも状況は同じである.状況を物質の密度ρ(r)のようなもので表せば
固体の場合は,状況を原子位置,あるいは電子密度分布と考えればよい.
3つのベクトルa, b, c を結晶格子の 並進ベクトル と呼ぶ.a, b, cによって構成される平行六面体を 単位胞 (unit cell) という( 図 4 ).また,ベクトルa, b, cの大きさ(長さ)を 格子定数(lattice constantまたはlattice parameter) という.結晶とは,ベクトルa, b, cで定まる単位胞をすき間なく3次元的に並べたものということができる.
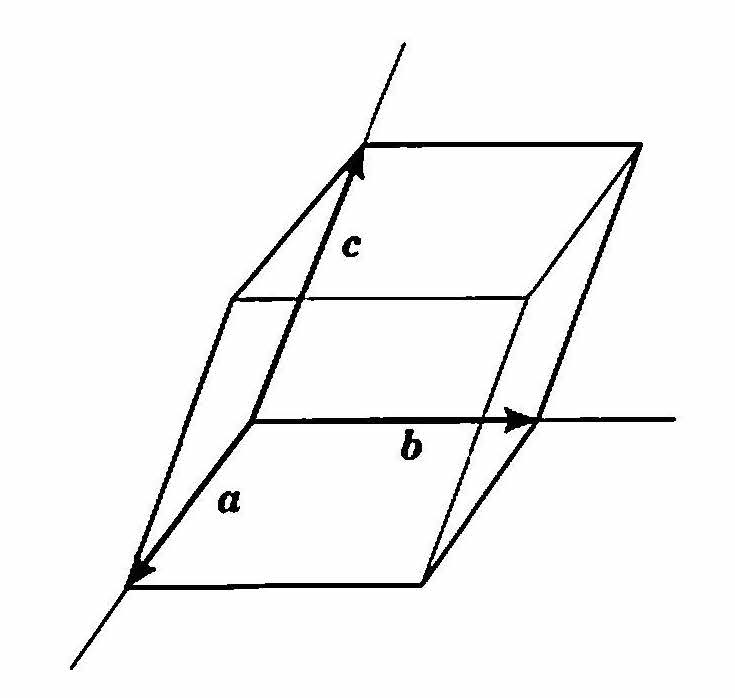
図 4 3つの並進ベクトルa, b, cによって構成される平行六面体(これが結晶格子の単位胞である)¶
結晶系とブラベー格子¶
さて,結晶は,単位胞の形によって7種類の 結晶系 に分けられる.さらに,そのなかの格子点の位置によって14種類の ブラベー格子(Bravais lattice) に分類できる. 格子点 とは,原子の位置と考えればわかりやすいが,その位置に原子がなくまわりの原子群を表すものでもよい.原子の種類や原子群の並べ方はすべての格子点において同一でなければならない.ブラベー格子の概念を用いれば,ある物質(完全結晶)の1 mol中6.02×1023個の原子配列は,単位胞とその各格子点における原子群を与えることで記述できる.
図 5 は7つの結晶系を系統的に分類し,各結晶系に属するブラベー格子を示したものである.左上の三斜晶は,3つの格子定数の長さが異なり(a≠b≠c),格子ベクトル間の角度も異なっていて (α≠β≠γ),最も対称性が低い構造である.矢印の順に対称性が高くなり,最も対称性の高い構造が立方晶(a=b=c かつ,α=β=γ=90°)である.六方晶および菱面体晶は別の系統で対称性が高い構造である.

図 5 7つの結晶系の系統図¶
金属の代表的な結晶構造¶
金属は結合が等方的であるため,前述のように原子をパチンコ球のように考えてよい.そうすると,球を最大限密につめ込んだ結晶構造が金属の構造として適すると考えられる. 図 6 に示すように,原子球を平面上に密に並べ,その上に原子を積んでゆく.原子はパチンコ球と違ってお互いに引きあっているのでくずれることはない.第1層の原子の中心位置を上からみてAで表す.そのすき間はBとCがある.第2層の原子をすき間Bに積むとす る.すると第3層の原子をすき間Cに積む方法とA原子の上に積む方法とがあり,異なったパッキング構造になる.前者は ABCABC… ,後者は ABABAB… の積み重ねとなる.
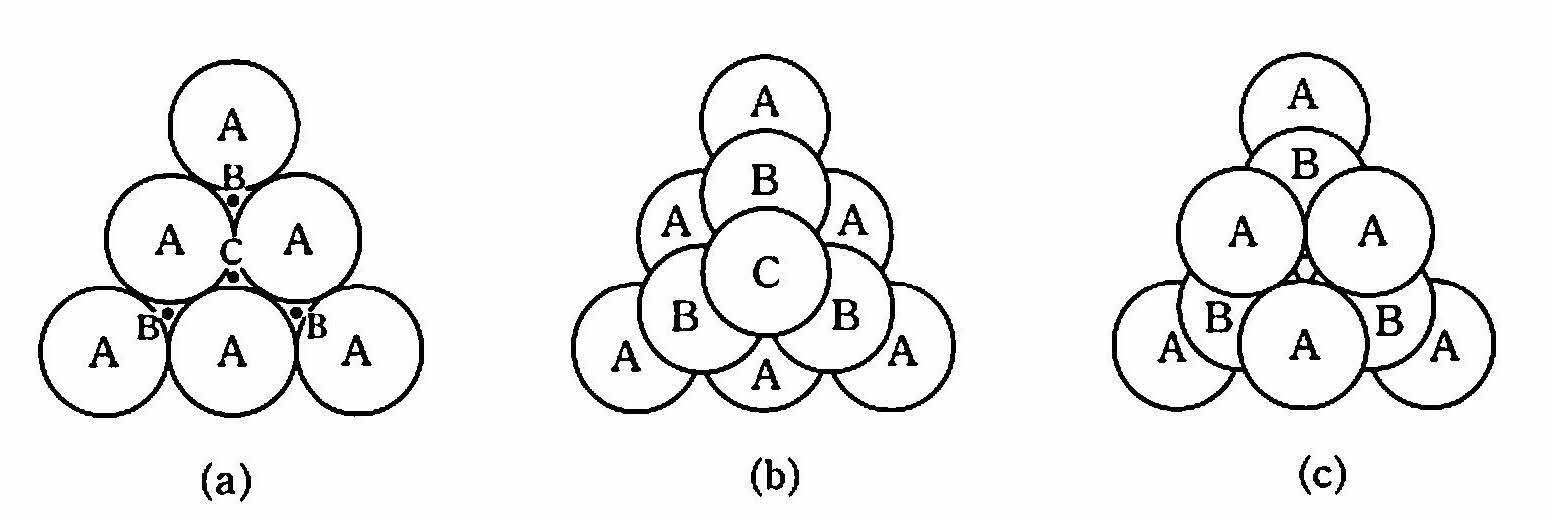
図 6 等しい大きさの球の最密充てん構造 (a)最密面 (b)面心立方格子 (c)最密六方格子¶
図 7 (a)は 面心立方格子 (face centered cubic lattice, FCC格子と略す) の原子配列を示す.これを対角線の方向 (〈l l l〉方向,後述)からみるとABCABC…の積み重ねになっている. 図 7 (b)に 最密六方格子(hexagonal close packed lattice, HCP) の原子配列を示す.底面に平行にABAB…の積み重ねになっている. 図 7 (c)は 体心立方格子(body centered cubic lattice, BCC) の原子配列を示したものである.この3つは金属の代表的な結晶構造である.面心立方構造をとる金属としては銅,ニッケル,アルミニウムなどがあり,最密六方構造は亜鉛やカドミウムなど,体心立方構造はα鉄(910℃でFCCのγ鉄に変態),バナジウム,タングステン,ナトリウムなどがある.
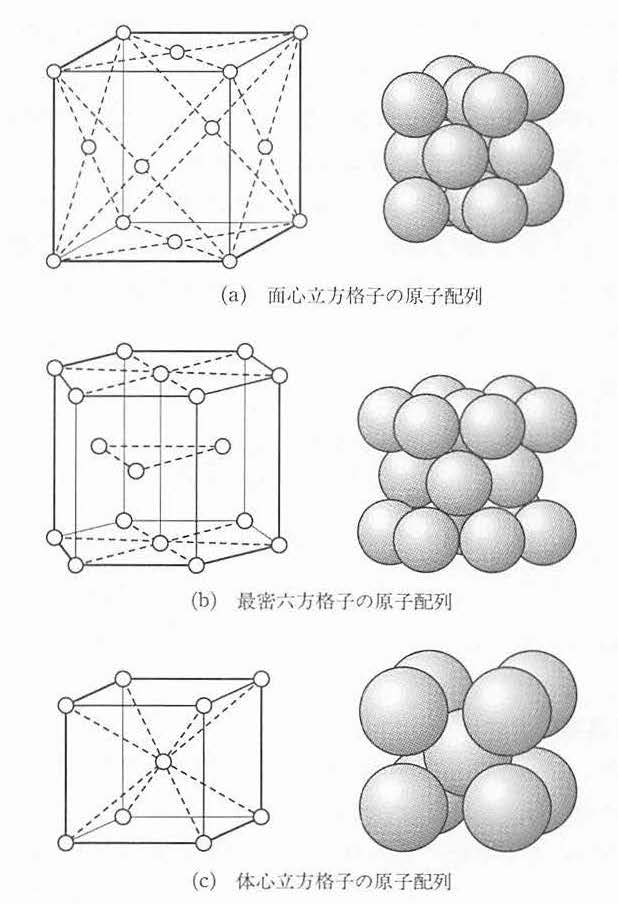
図 7 金属の代表的な結晶構造¶
次にこれらの結晶の 充てん率 を求めてみよう.充てん率とは, 単位胞中に含まれる原子球の体積を単位胞の体積で割ったものである .面心立方結晶は,単位胞中に4つの原子を含んでいる.すなわち,立方体の6つの面の中心におのおの原子があり,各原子は2つの単位胞に属しているので l/2×6=3個の原子が1つの単位胞に属している.また立方体の8つの頂点(隅)の原子はあわせて1/8×8=1個の原子が1つの単位胞に属している.合計3+1=4個となる.立方格子の辺の長さ(格子定数)をaとし,原子半径をrとすると \(4r = \sqrt{2}a\) の関係がある.ゆえに,充てん率は次のようになる.
最密六方結晶の充てん率もやや複雑だが同じように求まる.面心立方結晶と面の積み重ね方が違うだけなので,結果は同じく0.74である.
BCC結晶の充てん率が FCCやHCPに比べて低いのは,最密構造になることを妨げる理由があるからである.その理由としては,γ鉄のようなBCC金属には結合の異方性が多少含まれていること,熱振動によるエントロピー効果などが考えられる.
格子定数の値はどのくらいの大きさだろうか? 金属結晶の場合,1つの単位胞に含まれる原子の数はBCC格子(γ鉄など)で2個,FCC格子(Cuなど)で4個だから,格子定数は原子の大きさより少し大きい程度である.その正確な値は,後述するX線構造解析によって求めるが,結晶構造がわかっていれば,その物質の密度からほぼ正確な値を計算することができる.
例として,α鉄の格子定数を求めてみよう.α鉄の室温での密度ρはおよそ7.9 g/cm3である.鉄の原子量Mは55.85であり,これは鉄1 molが55.85 gであることを意味する.これより,鉄1molの体積(モル体積)はM/ρである.一方,アボガドロ数(1 mol 中の原子の数)N = 6.02×1023個/molを用いて,モル体積を表せば,Na3/2, ここでBCC 格子の単位胞には2 つの原子が含まれることを用いた.ゆえに
なお, X線回折法 によって求めた正確な値はa=0.2866 nmである.密度というマクロな量から,格子定数というミクロな最をかなり正確に知ることができる.
結晶学的記述¶
結晶を研究するうえで,結晶中の原子位置や方向,結晶中の原子面を記述する用語が必要である.それらはいずれも 単位胞 を基準として記述する.
まず原子位置を表す座標であるが,原点を(0,0,0)とし,x軸方向に格子定数aの長さ離れた位置を(1,0,0)とする 体心位置は \((\frac{1}{2},\frac{1}{2},\frac{1}{2})\) と表せる( 図 8 ).一般に,実長をx,y,zとしたとき,それを格子定数a,b,cで割った値u=x/a,v=y/b,w=z/cを用いて原子座標を (u,v,w) と表す.
結晶方位は,原点から点(u,v,w)への方向を [uvw] と表す.ただし,分数となる場合は整数を掛け,最小の整数の組合せとする.たとえば点 (1,0,0)への方向は[100],点 \((\frac{1}{2},\frac{1}{2},\frac{1}{2})\) への方向は[111]と表す.点 \((0,\frac{1}{2},1)\) への方向ならば[012]である.また,たとえば点(1,-1,0)への方向は数字の上にマイナスをつけて \([1\bar{1}0]\) と表す( 図 8 ).立方晶(a=b=c) では[110], [101], [011]および[110], [001], [011]など等価な方向をまとめて \(\langle 110\rangle\) のように表す.
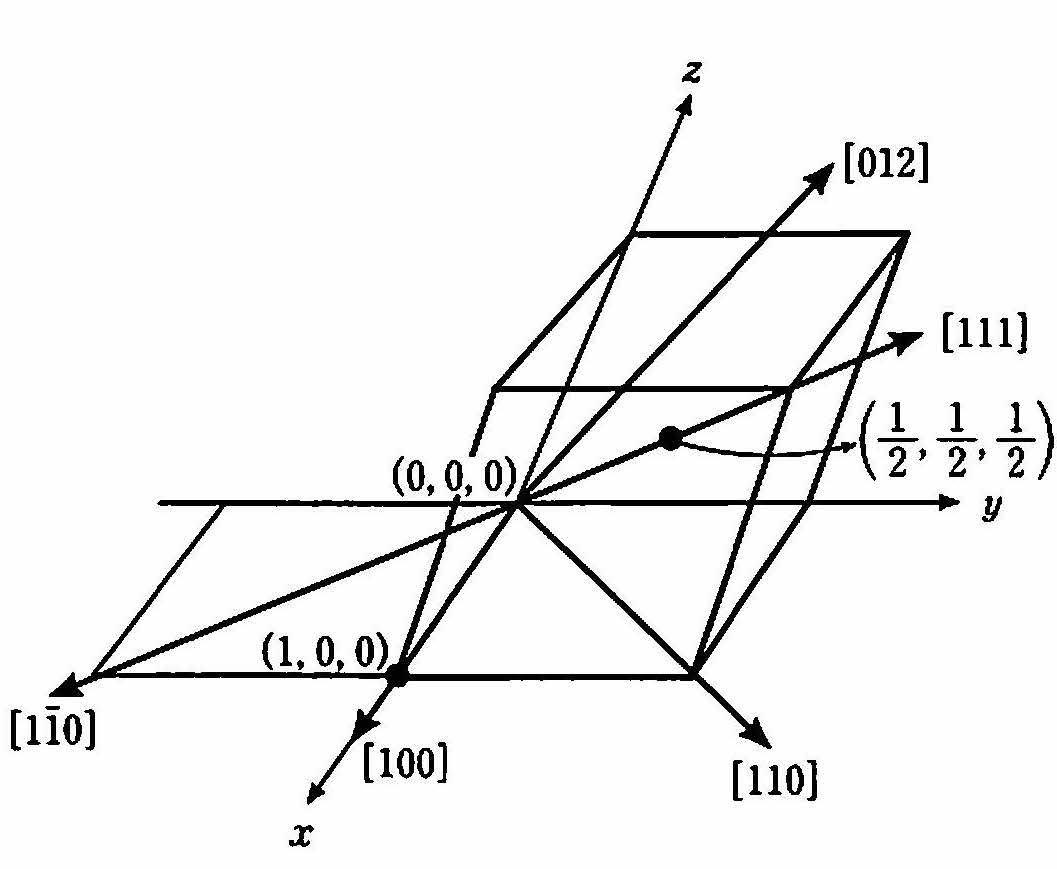
図 8 原子位置と結晶方位の表し方¶
次に,結晶中の原子面の表し方を説明しよう.ある原子面がx, y, z軸と交わる交点の座標を格子定数を単位として表し, その逆数をh, k, l とする.ただし,h, k, lが分数を含む場合には整数を掛けて,最小の整数の組合せとする.このときこの原子面を (hkl) と表示する.たとえば, 図 9 のように,点(1,0,0), (0,1/2,0),(0,0,1/3)でx, y, z軸を切る面は,(1,2/1,3/1) = (123)面と表示される.x軸を1/h, y軸を1/kで切り,z 軸に平行な面は,z軸と交わらないから(h,k,1/∞)=(hk0)面となる.このような結晶面の表示を ミラー指数(Miller index) 表示という.代表的な面を 図 10 に示した.立方晶の場合には,(110), (101), (011)および \((1\bar{1}0)\) , \((10\bar{1})\) , \((01\bar{1})\) の等価な6つの面をまとめて {110} のように表す.

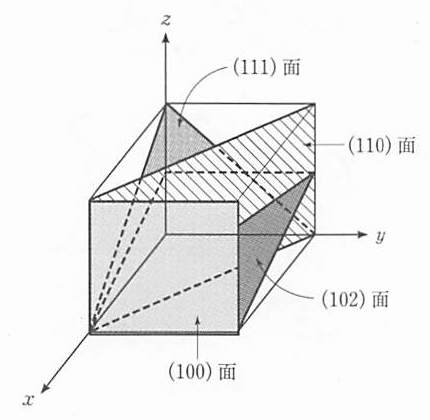
ある1つの単位胞の原点を基準として,たとえば(110)面を定義したとしよう.原点をずらして隣の単位胞についても同じように(110)面を定義することができる.この2つの面は平行である( 図 11 ).このようにして結晶中に無数の等間隔に並んだ (110)面を求めることができる.これらは同じミラー指数で表示される.すなわち,(hkl)面という表示は等間隔に並んだすべての平行な面を表している.
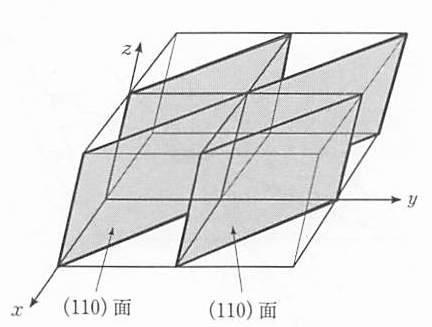
図 11 (hkl)という表示は.結晶中の等間隔に並んだ無数の(hkl)面を表す(図は(110)面の組を示す)¶
では,それらの面の間隔を求めてみよう.(hkl)面の面間隔をdhklと書く. 3つの並進ベクトルが直交 (α=β=90°)する斜方晶系や立方晶系の場合について考える. 図 12 に示すように(110)面の面間隔は,原点から(110)面への垂線の長さに等しい.a=bの場合は,ただちにd110= a/ \(\sqrt{2}\) である.a≠bの場合は垂線とx, y軸とのなす角をα1, α2として次のような関係がある.
直交座標系では \(\cos^2 \alpha_1 + \cos^2 \alpha_2=1\) であるから,
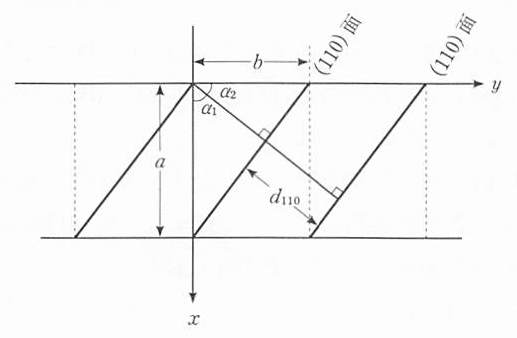
図 12 格子定数 a,bから(110)面の面間隔が求まる.¶
ゆえに
同様に(hkl)面の面間隔を次のように求めることができる。原点から(hkl)面へ降した垂線とx,y,z軸とのなす角をα1,α2,α3とすると,その垂線の長さは, 図 13 を参照して
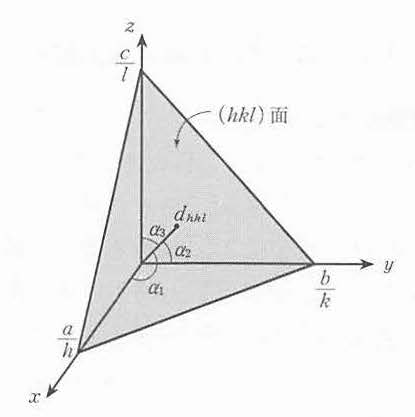
図 13 (hkl)面の面間隔dhklは原点からの垂線の長さに等しい.¶
斜方晶のような直交座標系では
であることを用いて
ゆえに
立方晶では,a=b=cとおいて
回折現象と結晶構造解析¶
X線回折現象とブラッグ条件¶
結晶が規則正しい原子の並びによって構成されていることが確認されたのは1912年のラウエ(Laue)によるX線回折斑点の発見とその解析によってである.ラウエはX線を照射し,背後に写真乾板を置いて観測したところ,規則正しい回折斑点を見いだした.光の回折と同じ原理で結晶の格子定数と同じ0.1 nmオーダーの波長をもつX線が回折現象を示したのである.ラウエ(独)につづいて翌年にはブラッグ父子(英),また1913-14年には寺田寅彦がX線回折の研究を行っている.
ラウエは理論的考察によって,X線回折の式(ラウエ条件またはラウエの法則と呼ばれる)を導いたが,ここでは,より直観的に理解しやすい ブラッグの法則(Bragg's law) または ブラッグ条件 と呼ばれるものについて説明しよう.X線が結晶に入射すると,原子との相互作用によって散乱され,それら異なった原子からの散乱波が強め合う条件のとき,その方向に回折波が観測される.いま,結晶中の(hkl)面に対し,角度θでX線が入射した場合を考えるとその第1層にある原子からの散乱波とその下の層の原子からの散乱波の位相がそろうのは, 図 14 に示すように,2つのビームの光路差が波長の整数倍になるときである.すなわち,
ここでdhklは(hkl)面の間隔,λはX線の波長である.これをブラッグの法則と呼ぶ.実際にはn=1のみを考えればよい.n=2以降は,dhkl/nの面間隔をもつ面,すなわち(nh,nk,nl)面からの回折と表示する.一般に面間隔の最も大きい面の回折が角度θの小さい内側に観測され,θが90°を超えるような面間隔の小さい面は観測されない.回折斑点の配列から結晶構造を求めることができ,また,いくつかの面についてθを精密に測定して格子定数を決定できる.
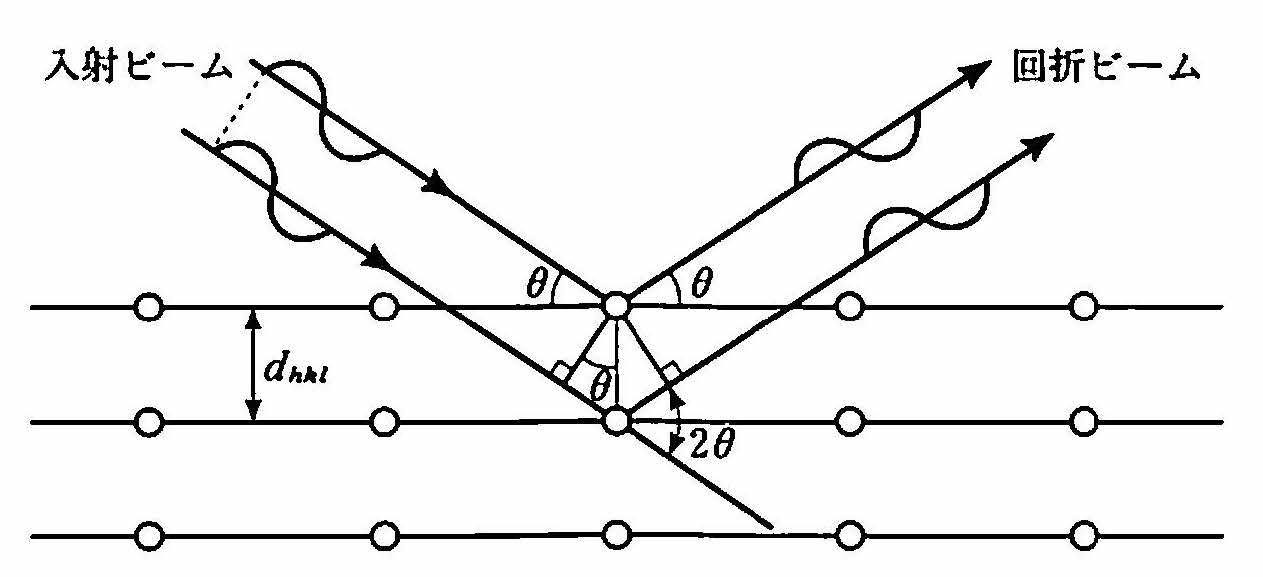
図 14 ブラッグの回折条件¶
X線ビームの回折はおのおのの原子によってなされるのに,結晶中の原子面による反射を考えるのはおかしいと思った読者もおられるだろう.X線ビームが平面波であるならば,同一原子面にある原子からの回折は同じ位相をもつと考えてよい.それゆえ,回折は原子面によって起こると考えてよく,面内の原子位置には依存しない.
物質の結晶構造を知り,格子定数を求めるのに現在最もよく用いられるX線回折技術はディフラクトメータ法と呼ばれるものである.これは 図 15 に示すように,平板に結晶粉末をつめたものを試料とする.平板試料と入射X線ビームとの角度をθとしたとき,角度2θの位置にX線検出器(比例計数管や半導体検出器など)を置き,θを時間とともに変化させてブラッグ回折ピークを観測する(これはしばらく前まで標準的であった粉末写真法と等価の測定法である.したがって古いX線回折の教科書で粉末写真法について書いてあることは,技術的問題は別として,ディフラクトメータ法にもあてはまる).
ディフラクトメータ法で粉末結晶を使う理由は何か.もし単結晶を用いたならば,この方法では特定の条件のときしかブラッグピークが観測されない.なぜならば,単結晶中の(hkl)面がブラッグ回折を起こすのは式 (12) を満たす特定のθのときであるが,そのとき(hkl)面がX線ビームに対し,θの方向を向いている保証は全くないからである.微細な粉末状の結晶を多数つめた試料では,結晶がランダムにあらゆる方向に向いていて,そのどれかの結晶粒が,ブラッグ条件を満たすθの方向を向いていてくれると考えるのである.回折線はある角度幅(1/100°程度)をもっているから,乳鉢ですりつぶした程度の粉末結晶(0.1 mm 程度)でこの条件は満たされる.

図 15 ディフラクトメータ法の概念図。試料がθ回転するときカウンターは2θの位置にくるように動く.図中のDS, SS, RSはスリット系である.¶
回折線出現の条件(消滅則と規則格子反射)¶
X線回折によって結晶の構造がわかるのは ,その回折波の現れる角度によって格子面間距離がわかるのみならず,結晶構造に依存して, 観測される回折波と消えてしまう回折波があるから である.
簡単な例として体心立方(BCC)格子の(001)面による回折を考えよう. 図 16 は(001)面を[100]方向(x軸方向)から見た図である.(001)面がブラッグ回折を起こす条件は式 (12) から
であるが,この角度θで入射したX線はその途中のd110/2の位置にある(002)面によっても回折され,その回折波は光路差がλ/2で,位相差が180°となり, (001)面の回折と打ち消しあう .よってBCC結晶では(001)面の回折は観測されない.どの回折ピークが欠けるかは結晶構造によって異なり,それを表す規則を 消滅則 という.BCC格子では,{100}, {111}, {210}面のようにh + k + l=奇数となるような格子面の回折線が消滅する.これらの面では,面の中間に等価な原子面が存在し,回折線の打消しあいが起こる.消滅則の一般ルールは次節で述べる結晶構造因子を用いて導くことができる.
次に, 図 16 において,中間の(002)面上の原子と(001)面上の原子とが異なる場合(たとえばFeとAl)はどうなるであろうか.Fe原子とAl原子とは,X線を散乱する強度が異なるので,2つの面からの回折線は完全には打ち消しあわず,弱められて残る.すなわち,消滅則によって消えた回折線が復活する.このようなことは,規則格子の形成によって起こる.たとえば,Fe原子とAl原子がランダムに配列したFe-Al合金はBCC構造をもち,(001)回折線は現れないが,Fe原子がBCC格子の隅である(0,0,0)位置を占め,Al原子が(1/2,1/2,1/2)位置を占めるような規則化(CsCl構造になる)が起こると,(001)ピークが観測されるようになる.よってこのピークを 規則格子回折線 あるいは 規則格子反射 (superlattice reflection) と呼ぶ.
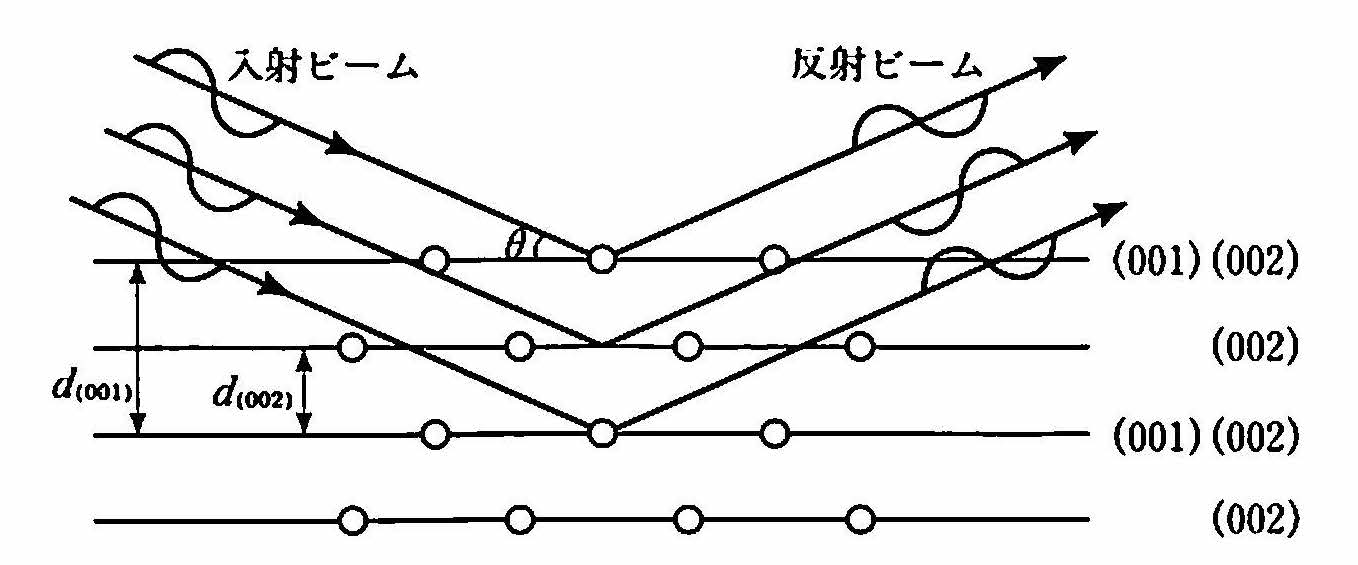
図 16 消滅則と規則格子回折¶
結晶構造因子¶
前節でBCC格子を例にとって消滅削や規則格子線について直観的に説明した.単位胞にいくつもの原子がある場合やもっと複雑な結晶格子の場合はどのように考えたらよいのだろうか.これから説明する結晶構造因子(crystal structure factor)という概念を用いれば,どのような場合にも答が導き出せる.
ある結晶の(hkl)面の結晶構造因子をFhklと書き,次の式によって定義する.
ここに, \(\Sigma_{j=1} ^n\) は1つの単位胞のなかのすべての原子について足し合わせることを意味し,fjはそのなかのj番目の原子の散乱の強さを表す.fjをその原子の原子散乱因子(atomic form factor)という.uj, vj, wjはj番目の原子の位置座標(原子座標)で,x,y,z軸の格子定数a,b,cを単位として表す.すなわち,実際の長さxj, yj, zjを格子定数で割った値で,uj= xj/a, vj=yj/b, wj= zj/cである.たとえば(1/2,1/2,1/2)の位置にある原子はuj= vj= wj= 1/2 である.なお,iは虚数単位である.この構造因子Fhklを計算すれば回折の様子がすべてわかる.以下,Fhklの意味を説明しよう.
まず,簡単のため(00l)面がブラッグ条件を満たすとき,点(000)と点(00z)との位相差φを求める. 図 17 に示すように,(00l)面の反射が強めあうというブラッグ条件は,ビーム1と2の光路差δ12が波長λに等しいことである.すなわち,
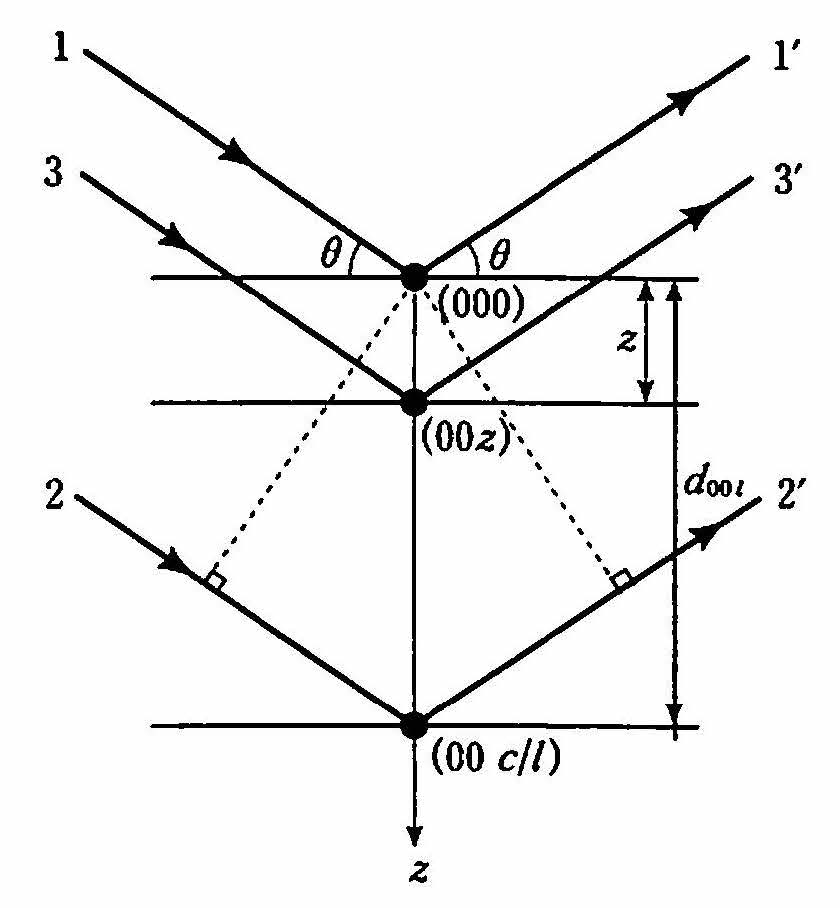
図 17 (00l)面の結晶構造因子F00lを求めるための図.(00l)面がブラッグ条件を満たすとき,すなわち点(000)と点(00 c/l)からの回折が強め合うとき,任意の点(00z)にある原子からの回折を計算する.¶
このとき点(00z)にある原子(●印)で回折されたビーム3の光路差を求めると,
d00l= c/l, w=z/cなる関係を用いると,
となる.位相差はδ12に対しφ12= 2πであるから,
一般に,(hkl)面がブラッグ条件にあるとき,原点(0,0,0)と点(x,y,z)とからの回折波の位相差は,上の場合と全く同じように導くことができ,
さて,ある位置での波の時間変化はA sin ωtと表すことができ,位相がφ進んでいる波はA sin(ωt+φ)と表せる.これを複素数表示すれば, \(A e^{i \omega t}\) および \(A e^{i(\omega t + \phi)}\) である.この2つの波の合成は
である.
以上の結果をもとに,単位胞中の1, 2, 3, …,j, …,n番目の原子からの回折波の合成は,
式 (15) を用いて,
となる.上式で時間で振動する因子 \(e^{i \omega t}\) を除いたものが構造因子Fhklである.すなわち,Fhklは単位胞中のすべての原子からの回折波を合成した波の振幅を表している.X線の回折強度は振幅の2乗に比例するから
ここで比例定数Kは,(hkl)反射の多重度m, 吸収因子A, 温度因子(デバイーワラー因子)T, 角度因子の積で,次式のように表される(詳しくはX線回折の教科書を参照されたい).
式 (14) を用いてα-Feの(hkl)面の構造因子を求めてみよう.α-FeはBCC構造だから原子位置は(0,0,0)と(1/2,1/2,1/2).ゆえに
BCCでは(110), (200), (211), (220)などの回折線が残る.
次に,FeAl規則格子の回折強度を求めよう.同様に
これより
h+k+lが奇数である回折線が規則格子線である.規則格子線では,異なる原子散乱因子の差の2乗に回折強度が比例するから,一般に強度が弱くなる.原子散乱因子fはその原子の電子の数,すなわち原子番号に比例するから,鉄とコバルトあるいは銅と亜鉛のように周期律表上で隣あった元素同士の場合は,非常に観測が困難になる.たとえば,原子番号29の銅と30の亜鉛の場合に,規則格子線と基本格子線との強度比は,比例定数Kの違いを無視すれば,およそ
となる.